علاج جيني يشفي طفلاً من مرض دم يصيب الملايين
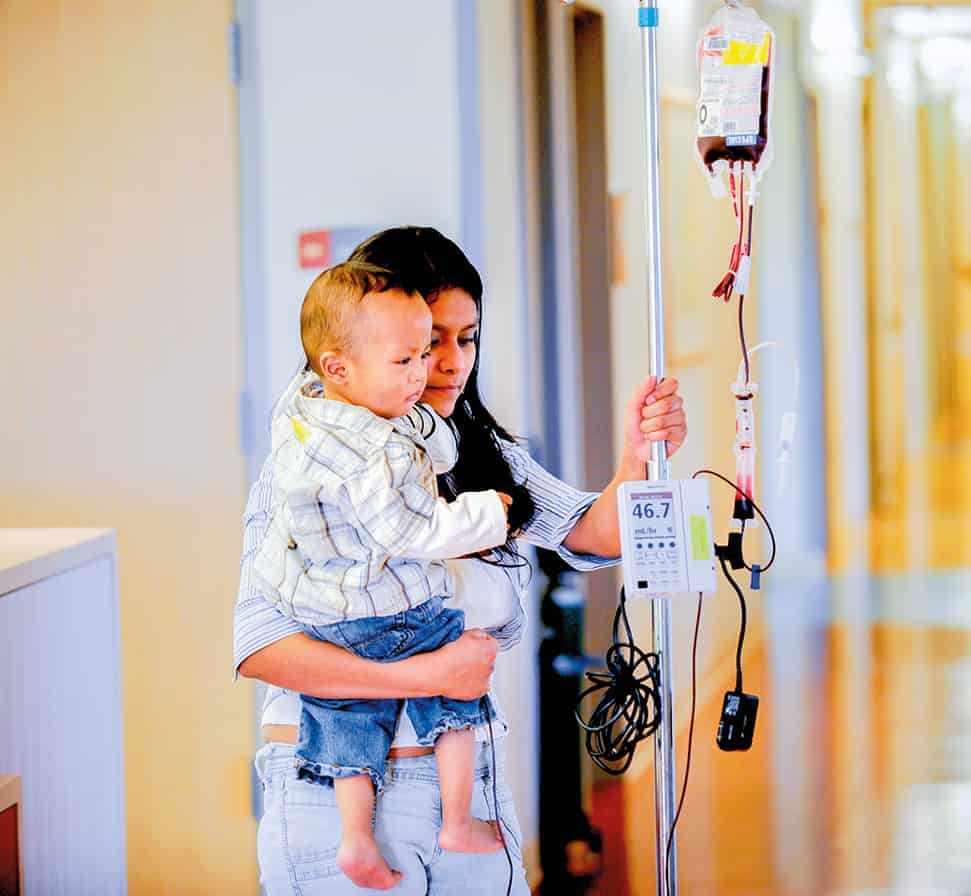
يبدو أن صبيّاً مراهقاً مصاباً بمرض يصيب الملايين حول العالم قد شُفِيَ بعد الخضوع للعلاج بالجينات، إذ يبدو أن العلاج قد أوقف الأعراض المؤلمة لفقر الدم المنجلي Sickle cell disease، مما يظهر إمكانية استخدام العلاج بالجينات لمعالجة الأمراض الوراثية الشائعة.
وفكرة العلاج بالجينات -التي تقوم على استخدام سلاسل الحمض النووي (الدنا) DNA للتعويض عن الجينات المعطلة عند الشخص- تعود تقريباً إلى ثلاثة عقود مضت، لكن المقاربة العلاجية تلك استخدمت منذ ذلك الحين فقط لعلاج الأمراض النادرة جداً (اقرأ: طريق النجاح الطويل).
وعلى العكس من ذلك، فإن مرض الخلايا المنجلية يصيب 100 ألف شخص في الولايات المتحدة وحدها، وإذا أثبت العلاج نجاحه في التجارب الأكبر والأوسع نطاقاً، فقد يؤدي ذلك إلى إدخال العلاج بالجينات إلى مجال الاستخدام واسع النطاق.
وتقول ديبورا جيل Deporah Gill من جامعة أوكسفورد: “قد يكون ذلك نقطة تغير كبيرة، وحقيقة كون الفريق لديه حالة مريض حصل على فائدة سريرية حقيقية، وبيانات حيوية تثبت ذلك، فإنه أمر مهم حقاً.”
تصنع أجسام الأشخاص المصابين بفقر الدم المنجلي نسخاً غير طبيعية من الهيموغلوبين Haemoglobin، وهو بروتين الدم الذي ينقل الأكسجين إلى أنحاء الجسم، وقد يحدث ذلك بسبب طفرات في الجين (المورثة) الذي يصنع وحدة من وحدات الهيموغلوبين تدعى بيتا-غلوبين Globin Beta، فتؤدي الطفرة إلى تجمع الهيموغلوبين معاً مما يشوّه شكل خلية الدم الحمراء إلى الشكل المنجلي الذي يجعلها تَعلَق في الأوعية الدموية داخل الجسم.
ويخضع الأشخاص المصابون بهذا الاضطراب لنقل الدم لتصفية الانسدادات المؤلمة ومنع حدوث انسدادات أخرى، ويمكن علاج هذا المرض بزرع نخاع العظم، لكن لا يمكن العثور على متبرعين مطابقين إلا لنحو 10 % من الأشخاص المصابين بهذه الحالة.
أما الآن، فيبدو أن فريقاً في فرنسا طوّر علاجاً قد يكون فعالاً لكل شخص مصاب بهذا الاضطراب. ففي البداية، أخذ الفريق الخلايا الجذعية من نخاع العظم من الصبي عندما كان عمره 13 عاماً، وزرعوا في تلك الخلايا نسخاً إضافية طافرة من الجين الذي يرمّز لبيتا-غلوبين، إذ صُمّمت تلك النسخ لصنع بيتا-غلوبين يتداخل مع عمل بروتينات الصبي المَعيبة، مما يمنع تجمعها معاً.
أعاد الباحثون تلك الخلايا الجذعية إلى جسم الصبي، وبعد مضي نحو ثلاثة أشهر، بدأ بإنتاج كميات كبيرة من الهيموغلوبين ذي السلوك الطبيعي (New England Journal of Medicine, DOI: 10.1056/NEJMoa1609677).
وتقول قائدة الفريق مارينا كافازانا Marina Cavazzana من مستشفى نيكر للأطفال Necker Children’s Hospital في باريس: “عمر هذا المريض الآن 15 عاماً وهو لا يتعاطى أي دواء من أدويته السابقة، وهو لا يعاني الألمَ الذي كانت تسببه انسدادات الأوعية الدموية، كما توقف أيضاً عن تناول المسكنات الأفيونية.”
وكافازانا واثقة بأن هذه الفوائد ستدوم، وتتابع قائلة: “كل الفحوص التي أجريناها على دمه تظهر أنه شُفِي، لكن الحصول على النتيجة الأكيدة يأتي فقط من المراقبة والمتابعة طويلة الأمد.” كما تقول إنّ فريقها قد عالج سبعة مرضى آخرين يظهرون تطوراً واعداً.
ويقول جون تيزديل John Tisdale من معهد الولايات المتحدة الوطني للقلب والرئة والدم US National Heart, Lung, and Blood Institute في ماريلاند: “إننا متحمسون جداً لنتائج هذا العمل، ويقدم هذا النجاح دعماً لهذه الاستراتيجية والاستراتيجيات الأخرى التي تستهدف هذا الداء الخبيث.”
أما ديفيد ويليامز David Williams من مستشفى بوسطن للأطفال Boston Children’s Hospital في ماساتشوستس فيقترح أن الصبي قد يتعرض في بعض الأحيان لانسدادات، لأن جيناته الأصلية ما زالت قادرة على إنتاج هيموغلوبين معيب، إذ يقول: “من المهم أن نرى ما يحصل بمرور الزمن، وما عدد المرضى الآخرين الذين سيجنون فوائد مماثلة.”
لكن، إذا أثبت العلاج الجيني فاعليته في التجارب الأكبر، فقد تؤدي تكلفته إلى حصر استخدامه في البلدان الغنية.
ويقول ستيوارت أوركين Stuart Orkin من جامعة هارفارد للطب Harvard Medical School: “يجب أن نكون واقعيين ونتذكر أن هناك مئات الآلاف من المرضى المصابين بفقر الدم المنجلي في الدول الأقل تطوراً، وأن إمكانيةَ استيراد العلاج وتبنيه قليلةٌ في الدول ذات الأنظمة الصحية ضعيفة التطور.”